apert syndrome
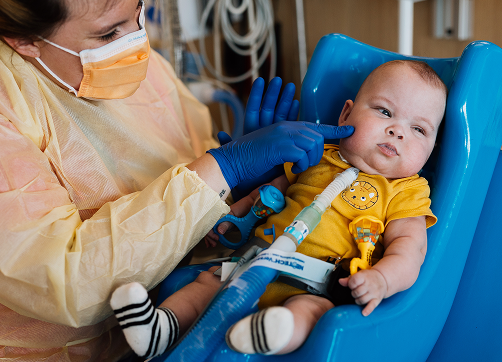

about condition
Apert syndrome is a genetic congenital condition characterized by skeletal malformations. Severity of symptoms varies among individuals. A key feature of Apert is the premature closure of the skull bones, called craniosynostosis. In addition, a varied number of fingers and toes may be webbed/fused together, called syndactyly. Premature fusion of the skull bones prevents the head from growing as it should, which leads to a sunken appearance in the middle of the face (midface hypoplasia), a beaked nose, a wrinkled forehead, and an opening in the roof of the mouth cleft palate). In individuals with Apert syndrome, an underdeveloped upper jaw can lead to dental problems, such as missing teeth, irregular tooth enamel and crowded teeth.
characteristics of this syndrome may include:
- Back of skull may be flattened
- Flattened nose with low bridge
- Webbed or fused fingers and toes
- Pointed top of head (acrocephaly)
- Late-closing skull ‘soft spot’
- Skull bone malformations (craniosynostosis)
- Intellectual disabilities are possible
- Possible breathing and swallowing issues
- Possible hearing loss, chronic ear infections
- High and broad forehead, wrinkled forehead
- Most often an opening in roof of mouth (cleft palate)
- Dental issues such as teeth delay, dental crowding, open bite
- Midface and upper jaw malformations (may require surgical intervention)
- Widely spaced eyes (hypertelorism), bulging eyes, down-slanting palpebral fissures (area between the open eyelids)
- Possible abnormal fluid accumulation in cavities of the brain (hydrocephalus)
- Heart malformations that may include: Hole(s) in the ventricular wall, aorta malformation
- Abdomen issues may include: Narrower opening between lower stomach and upper part of small intestine, esophagus blockage
- Other skeletal issues may cause: Short stature, fusion of the following: neck vertebrae, particular arm bones, wrist bones
- Kidney/Urinary Systems and Sexual Organ malformations may include: Enlarged kidneys due to blockage, out of position anus, vagina blockage, failure of testicles to fall
- Neurologic issues may cause: Varying degrees of developmental delay, mild to moderate intellectual disability (based on skull surgery age or any other brain malformations), enlarged brain cavity, other brain and skull malformations
*Surgical intervention may include cranial vault and midface distraction procedures to improve functions early on, as well as other necessary procedures as the child grows, such as hand and foot surgeries.
how do I know if my child will be born with Apert syndrome?
Sometimes when an ultrasound is performed during pregnancy, a doctor may notice non-typical signs which require additional testing, such as more in-depth testing with 2D or 3D ultrasounds or a fetal MRI, which these can reveal much more. After birth, clinical examinations, CT scans, MRI and other testing can help to diagnose the syndrome and any organ issues, such as kidneys, heart, and more. Genetic counseling is highly recommended.
how do you treat Apert syndrome?
The treatment of Apert is focused on the specific symptoms apparent in each individual since the symptoms can vary widely. Several specialties and physicians may be involved throughout the child’s life, such as craniofacial and reconstructive surgeries to correct skull and head malformations. Also surgeries to correct polydactyly and syndactyly, and reconstruction of other skeletal malformations. Patients may see some or all of the following physician specialties: cardiologist, otolaryngologist (ENT), audiologist, neurologist/neurosurgeon, ophthalmologist, dentist, orthodontist, oral surgeon and more. Early intervention may be important to ensure that children with this syndrome reach their full potential. Special services like physical and occupational therapies along with special education may be very beneficial.
FAQs
Apert syndrome affects an estimated 1 in 65,000 to 88,000 newborns. It appears males and females are affected in equal numbers. Over 300 cases have been reported since it was discovered in 1894 and 1906. Research indicates that Asian individuals have the highest incidence of this syndrome.
Yes, Apert is a congenital syndrome apparent at birth. This syndrome is thought to be a sporadic event (new genetic mutation change) and not inherited. Therefore, the chance of passing it on to future children would be rare, although not non-existent. (Only a few cases have been reported to be a dominant genetic occurrence where the chance of passing that on to future children would be a 50% chance for each pregnancy.)
Even though surgeries may often be in their future, individuals with Apert can have a typical life span. (There are incidences of children with severe heart issues that may affect their life span.)
The gene involved with this syndrome is FGFR2. (This gene is also known for causing similar syndromes, such as: Pfeiffer, Crouzon and Jackson-Weiss syndromes.)
– Mutations from this gene known for causing Apert syndrome are: Ser252Trp and Pro253Arg
The gene that causes Apert to occur plays an important and critical role in skeletal development. Genes give instructions for creating the proteins that play vital roles in our body. When a change/mutation in a gene happens, the protein that is important for building structures is disrupted. When a genetic change happens in FGFR2, it results in these receptors not communicating properly with fibroblast growth factors. This means the sutures in the brain do not form as they should, so consequently, this can disrupt the development of many other structures in the body. Since two different gene mutation changes are known for causing Apert, they can cause different malformations; therefore, varying in presentation.
Apert is part of a group type known as ACS1; all forms of ACS have craniosynostosis which affects growth of the skull and head.
Syndromes that are similar and related with Apert:
here when you need us
Whether you’re looking for the right provider, ready to make an appointment, or need care right now—we’re here to help you take the next step with confidence.
